

Estudos revelam que diabetes entre outras condições de saúde podem levar a uma Deficiência de G6PD adquirida, quando o individuo não possui mutação no gene G6PD, mas apresenta a deficiência de enzima de G6PD.
Vamos entender melhor no artigo cientifico de hoje.
Deficiência adquirida de glicose-6-fosfato desidrogenase
A deficiência de glicose-6-fosfato desidrogenase (G6PD) é uma condição hereditária causada por mutações no cromossomo X e é transmitida por herança ligada ao sexo. No entanto, o comprometimento da atividade da G6PD pode resultar de mecanismos bioquímicos que são capazes de inibir a enzima em condições clínicas específicas na ausência de um defeito estrutural no nível do gene. Nesta revisão narrativa, foram examinados vários cenários clínicos associados a uma deficiência “adquirida” de G6PD, fenotipicamente indistinguível da deficiência primária, bem como os mecanismos envolvidos. Hiperaldosteronismo e diabetes são os culpados mais comuns da deficiência adquirida de G6PD. Condições endócrinas e metabólicas adicionais podem causar deficiência de G6PD em pacientes hospitalizados e ambulatoriais. Ao contrário do defeito hereditário, a deficiência adquirida de G6PD é uma condição potencialmente curável pela remoção do fator responsável pela inibição enzimática. A conscientização sobre a deficiência adquirida de G6PD pelos médicos pode resultar em melhor reconhecimento e tratamento.
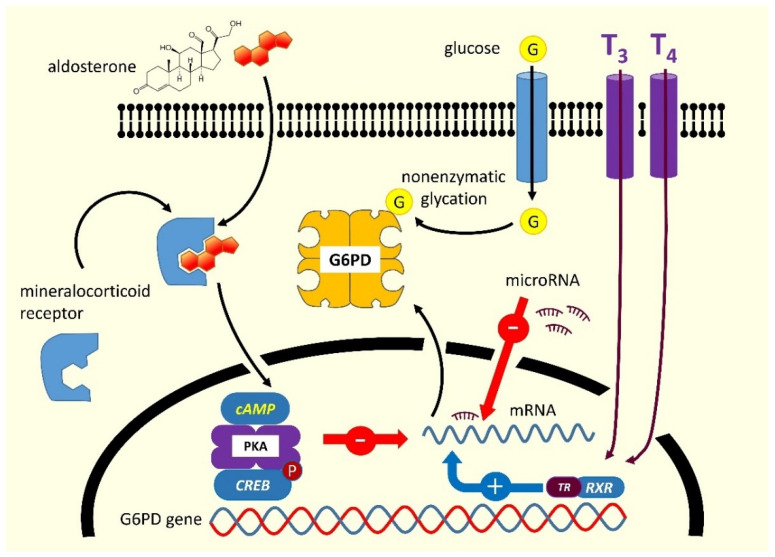
figura 1
Regulação da atividade de G6PD e potenciais mecanismos inibitórios. A aldosterona regula negativamente a expressão do mRNA de G6PD através da estimulação da síntese de adenosina monofosfato cíclica (cAMP), que se liga à forma tetramérica da Proteína Quinase A (PKA). A PKA fosforila a proteína de ligação ao elemento de resposta do fator de transcrição cAMP (CREB), bloqueando assim a transcrição do gene G6PD. Os hormônios tireoidianos ligam-se ao receptor tireoidiano (TR), formando um heterodímero com o receptor retinoide (RXR) e ativando a transcrição do mRNA da G6PD.
Além do envolvimento na hemólise aguda, a deficiência de G6PD foi recentemente reavaliada como fator de risco para um espectro mais amplo de doenças com patogênese inflamatória, como doenças cardiovasculares [11,12], asma [13] ou doença celíaca [14] , provavelmente decorrente da ativação do inflamassoma [15]. A maioria dos casos de deficiência de G6PD são distúrbios primários não modificáveis geneticamente transmitidos, e seu reconhecimento imediato pode evitar a exposição dos portadores a agentes potencialmente hemolíticos. Por outro lado, foram descritas formas adquiridas de deficiência de G6PD, geralmente transitórias e não relacionadas a defeitos genéticos [16,17,18]. Semelhante aos pacientes com deficiência hereditária de G6PD, aqueles com formas adquiridas são vulneráveis ao dano oxidativo, que geralmente é de curta duração e tratável removendo a causa subjacente. Os médicos devem reconhecer a existência e a frequência não negligenciável dessas formas adquiridas de eficiência reduzida da G6PD para fornecer terapia apropriada aos pacientes.
1 Introdução
A deficiência de glicose-6-fosfato desidrogenase (G6PD) é o distúrbio enzimático humano mais comum em todo o mundo, ocorrendo com mais frequência em áreas endêmicas de malária [1]. Esta condição é responsável por icterícia neonatal e/ou anemia hemolítica após exposição a uma série de drogas, infecções ou plantas contendo agentes oxidantes [2]. Na maioria dos casos, o distúrbio é herdado devido a mutações no gene que codifica G6PD (OMIM 305900), mapeado na região telomérica do braço distal do cromossomo X e transmitido por um mecanismo ligado ao sexo [1,3,4] . A enzima G6PD humana é encontrada no citoplasma de todas as células. Catalisa a primeira reação da via das pentoses-fosfato, fornecendo assim a ribose-5-fosfato necessária para a síntese de DNA, e o dinucleotídeo fosfato de nicotinamida adenina (NADP+/NADPH), principal doador de hidrogênio nas reações biossintéticas [2]. A enzima está envolvida na manutenção do nível de glutationa intracelular e contribui para neutralizar as espécies reativas de oxigênio [5]. Embora a G6PD possa ser geralmente encontrada em todos os tecidos, sua deficiência se manifesta essencialmente nas hemácias, onde, diferentemente de outras células nucleadas do corpo, nenhuma via bioquímica alternativa pode garantir a produção de NADPH. A G6PD existe como um monômero, dímero e tetrâmero, mas apenas o dímero é cataliticamente funcional, e a interconversão entre as três formas é crítica para a atividade da enzima [6]. Cada subunidade tem um sítio de ligação de glicose-6-fosfato, um sítio catalítico de NADP+ e um sítio de ligação alostérico de NADP+. Qualquer alteração desses domínios estruturais ou regulatórios, induzida por mutações genéticas ou na ausência delas por ligação com agentes externos pode modificar a eficiência da catálise, em alguns casos diminuindo-a severamente. Além disso, a influência de fatores endócrinos que reduzem a transcrição do gene G6PD é capaz de determinar uma forma adquirida de diminuição da atividade da G6PD [7,8]. Os mineralocorticoides diminuem a expressão do mRNA de G6PD estimulando a síntese de adenosina monofosfato cíclica (cAMP), que se liga à forma tetramérica da proteína quinase A; o último fosforila a proteína de ligação do elemento de resposta do fator de transcrição cAMP (CREB), bloqueando assim a transcrição do gene G6PD. Os hormônios tireoidianos ligam-se ao receptor tireoidiano (TR), que forma um heterodímero com o receptor retinoide (RXR), ativando assim a transcrição do mRNA da G6PD. Finalmente, G6PD é um gene alvo de vários microRNAs que desempenham um papel importante na regulação enzimática atuando principalmente pela supressão da tradução do mRNA de G6PD [9,10] (Figura 1).
2. Fatores que induzem potencialmente a deficiência de G6PD adquirida
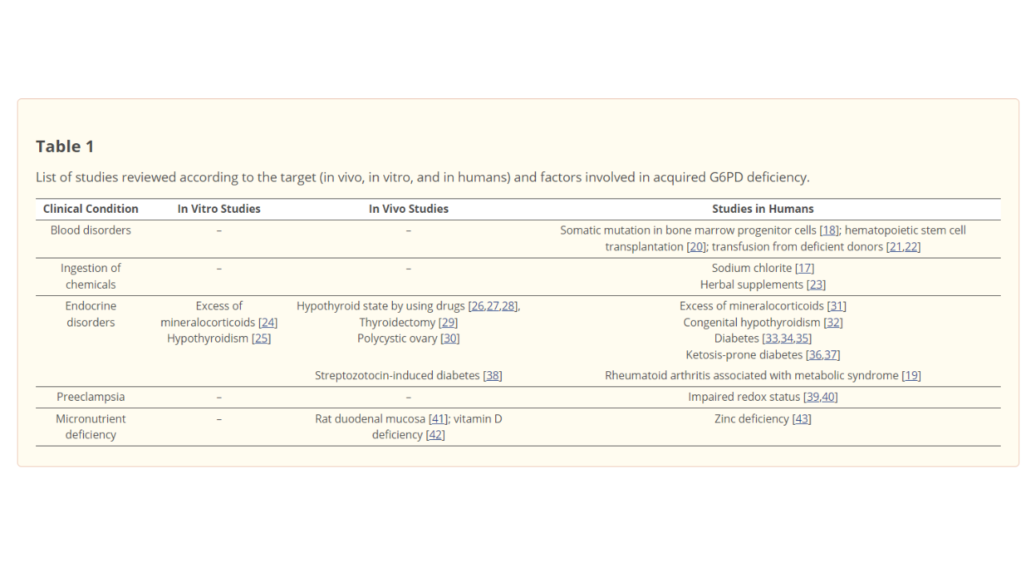
As formas adquiridas de comprometimento da G6PD podem ser difíceis de diagnosticar, devido à necessidade de descartar a presença de mutações na estrutura do gene. De fato, as formas adquiridas genuínas são aquelas em que os mecanismos bioquímicos produzem uma molécula enzimática intacta que é menos eficiente por diferentes vias. Assim, não é surpreendente que na literatura sejam descritos poucos casos de formas adquiridas de atividade reduzida da G6PD, uma vez que, na maioria das vezes, a presença de mutações genéticas não é rotineiramente rastreada nem definitivamente excluída [19]. De fato, alguns dos relatos de casos em que a deficiência foi testada no sangue e nenhuma mutação foi encontrada podem ser explicados por limitações técnicas, incluindo a falta de uma sequência completa do gene G6PD. Na seção abaixo, o comprometimento enzimático decorrente de mecanismos extrínsecos não genéticos relatados na literatura e resumidos na Tabela 1 é discutido criticamente.
tabela 1
Lista de estudos revisados de acordo com o alvo (in vivo, in vitro e em humanos) e fatores envolvidos na deficiência adquirida de G6PD.
2.1. Doenças sanguíneas
Estudos iniciais em grandes coortes de pacientes com distúrbios sanguíneos descreveram formas adquiridas de enzimopatias, particularmente deficiência de G6PD, com uma frequência de quase 3% [44]. A deficiência enzimática, aparentemente não relacionada a qualquer mutação demonstrável do gene G6PD e detectável na maioria dos tecidos, foi descrita em alguns relatos de caso. Recentemente, em um paciente líbio com leucemia mielomonocítica crônica, anemia hemolítica persistente foi observada na ausência de causas extracorpusculares óbvias [18]. Apesar de uma atividade reduzida de G6PD (21,3% de atividade residual), a sequência de DNA genômico, obtida do bulbo capilar, não revelou a presença de mutações. Em contraste, a sequência de DNA dos glóbulos brancos revelou uma nova variante sem sentido prevista para gerar uma proteína truncada não funcional. Esta deficiência de G6PD “adquirida” não foi o resultado de mutações genômicas no DNA, mas sim uma mutação somática surgindo em um clone específico de células da medula sanguínea [18]. Da mesma forma, pode-se presumir que pacientes que receberam células-tronco deficientes em G6PD de doadores inconscientes e subsequentemente considerados enzimopênicos desenvolveram uma forma adquirida de deficiência de G6PD. Um caso relatado por Au et al. sublinhou a importância de monitorar cuidadosamente a integridade do gene G6PD em potenciais doadores de transplante de células-tronco hematopoiéticas, bem como receptores que devem receber a mesma atenção médica que indivíduos com deficiência primária de G6PD [20]. Raramente, uma forma adquirida de deficiência de G6PD pode originar-se de transfusões de hemácias deficientes de doadores não previamente rastreados para a atividade enzimática [21,22]. Em alguns casos, a redução da atividade pode ser tão grande, ou o volume de sangue transfundido tão grande, que causa hemólise clinicamente significativa no receptor. Em populações com alta prevalência de deficiência de G6PD, é aconselhável avaliar G6PD em potenciais doadores de sangue, de acordo com as diretrizes da OMS. Doadores deficientes devem ser desencorajados a doar sangue. Além disso, bolsas de sangue contendo glóbulos vermelhos deficientes em G6PD não devem ser usadas para transfusão intrauterina, transfusão de troca neonatal ou para pacientes deficientes em G6PD [45].
2.2. Ingestão de Produtos Químicos
É bem conhecido que a ingestão de várias substâncias, produtos químicos ou medicamentos é potencialmente capaz de provocar uma crise hemolítica em indivíduos com um defeito predisponente da G6PD. No entanto, também há evidências de que várias moléculas podem induzir uma redução significativa na atividade catalítica da enzima sem evidência de transmissão genética de mutações G6PD [18,46]. Seu diagnóstico é desafiador, pois as mutações G6PD não são rastreadas rotineiramente, exceto em algumas populações com alta frequência do distúrbio [47]. No entanto, relatos de casos foram publicados descrevendo episódios hemolíticos tratados com sucesso sem redução significativa na atividade de G6PD no sangue, levantando assim a suspeita de uma forma adquirida transitória [18,44,48,49]. A ingestão de substâncias químicas específicas pode interromper significativamente a atividade da G6PD e, raramente, causar hemólise maciça. Em alguns desses casos, o culpado químico pode ter desencadeado uma predisposição genética latente à insuficiência enzimática, causando comprometimento da defesa antioxidante, notadamente nas membranas eritrocitárias. Ainda, outros casos podem surgir devido à ação direta do agente químico sobre a proteína enzimática. Um relatório de Hulshof et al. descreveram um caso de intoxicação por clorito de sódio, que causou hemólise grave na ausência de mutações predisponentes da G6PD [17]. Esta situação foi associada com metemoglobinemia grave que parecia ser um componente chave na fisiopatologia. O caso descrito foi tratado com a administração de azul de metileno, que tem efeito redutor da metemoglobinemia e também deve ser considerado como coadjuvante em casos semelhantes de deficiência adquirida de G6PD. Apesar de sua raridade, esses casos sugerem que em pacientes com anemia hemolítica, além dos exames laboratoriais comuns, é obrigatório coletar uma história medicamentosa abrangente, incluindo o uso de suplementos fitoterápicos e fitoterápicos da medicina tradicional chinesa e ayurvédica [23].
2.3. Distúrbios Endócrinos
2.3.1. Excesso de Mineralocorticóides
A inibição da G6PD de mamíferos por cetoesteróides C17 e C20 é conhecida desde o início dos anos 1960 [24]. Em contraste, os esteróides que possuem um grupo hidroxila em vez de um grupo cetona em C17 ou C20, como corticosteróides, estrogênio e progesterona, têm pouco ou nenhum efeito inibitório na G6PD [24]. Foi demonstrado que a inibição por cetosteróides é incompetitiva, o que é uma ocorrência rara no caso de substâncias diferentes dos substratos da enzima [50]. In vitro, a deficiência adquirida de G6PD foi descrita em associação com o excesso de precursores de cetosteróides secretados pela glândula adrenal [51]. A desidroepiandrosterona (DHEA, também conhecida como androstenolona, 3β-hidroxiandrost-5-en-17-ona ou 5-androsten-3β-of-17-one) é um importante hormônio esteróide endógeno [52]. Essa molécula e seu conjugado sulfato (DHEAS) apresentam a maior concentração sérica entre os andrógenos. O DHEA é um potente inibidor não competitivo da atividade da G6PD [53,54]. Como sua ação é dirigida principalmente aos eritrócitos saudáveis, reduz a glutationa corpuscular, dificultando a parasitização por Plasmodium falciparum [55] e, por isso, tem sido usada no tratamento da malária [56]. O excesso de andrógenos adrenais na patogênese da deficiência adquirida de G6PD também foi estudado em modelos animais de síndrome dos ovários policísticos [30]. A aldosterona, um hormônio adrenal estruturalmente semelhante ao DHEA, é um potente inibidor da atividade da G6PD. O hiperaldosteronismo e outras condições relacionadas ao excesso de mineralocorticóides promovem o estresse oxidativo e prejudicam a função endotelial e a reatividade vascular ao diminuir a atividade da G6PD [31,51]. Pelo contrário, a transferência de genes de G6PD em modelos animais melhora a reatividade vascular pela superexpressão de G6PD, confirmando assim a conexão patogenética acima mencionada. O mecanismo pelo qual a aldosterona inibe a G6PD tem sido investigado em modelos in vitro e in vivo, principalmente em células endoteliais. O hormônio aumenta os níveis de cAMP, que interage com a forma tetramérica da proteína quinase A (PKA), dissociando suas subunidades reguladoras das catalíticas. A PKA, por sua vez, inibe a expressão dos fatores de transcrição CREB/CREM que estão diretamente envolvidos na transcrição da G6PD [51] (Figura 1). De acordo com esses achados, foi levantada a hipótese de que uma forma adquirida e reversível de deficiência de G6PD pode ser induzida pelo excesso de mineralocorticóides, prejudicando assim a defesa antioxidante das células. Essa hipótese foi confirmada pela restauração da reatividade vascular normal, removendo o estado de deficiência de G6PD por meio do bloqueio do receptor de mineralocorticosteroide [20]. Além disso, uma vez que o uso de tióis naturais, como o ácido α-lipóico, demonstrou ser benéfico no tratamento de indivíduos com deficiência congênita de G6PD [57], é provável que a suplementação com este ou compostos semelhantes possa ser útil para equilibrar o status redox em indivíduos com deficiência adquirida de G6PD também.
2.3.2. hipotireoidismo
Modelos in vitro mostraram que a tiroxina (3,3′,5,5′-tetraiodotironina – T4), principal produto da glândula tireoide, pode ativar a enzima G6PD por competição com NADPH pelo mesmo sítio de ligação, apesar da falta de semelhança estrutural entre as duas moléculas [25,26]. Modelos in vivo também forneceram evidências de inibição de G6PD após diminuição da função tireoidiana. Por exemplo, em ratos tornados hipotireoidianos por meio de um tratamento combinado com propiltiouracil e iopamida, a atividade da G6PD foi reduzida em 28%, enquanto a administração de T2 restaurou a atividade [27,28]. A administração de 3,3′,5′-triiodotironina (T3) reverteu os efeitos em ratos tireoidectomizados. Em outro estudo realizado em roedores, uma diminuição significativa da atividade da G6PD foi observada após a tireoidectomia, enquanto o hipertireoidismo aumentou ambas as enzimas [29]. Esses achados confirmam ainda mais os efeitos hipermetabólicos dos hormônios tireoidianos no metabolismo celular. Embora a maioria dos efeitos fisiológicos dos hormônios tireoidianos seja mediada pela família c-erbA de receptores nucleares, há evidências de que os hormônios tireoidianos podem regular várias atividades enzimáticas independentemente da síntese de proteínas por meio de mecanismos não nucleares. Esse efeito tem sido investigado principalmente no tecido hepático, mas também foi observado no tecido adiposo marrom, que é o de maior potencial lipogênico do organismo [29]. A diminuição da atividade da função tireoidiana é biologicamente relevante e produz uma inibição significativa da G6PD e, por sua vez, uma maior vulnerabilidade do tecido a estressores [58]. Uma função tireoidiana adequada pode proteger contra o estresse oxidativo [59]. Em humanos, os achados são menos comuns, mas a deficiência de G6PD associada ao hipotireoidismo congênito foi descrita em recém-nascidos iranianos [32]. Curiosamente, os níveis de G6PD voltaram ao normal após 120 dias de tratamento com levotiroxina, indicando que a deficiência não era de origem genética.
2.3.3. Diabetes
Em animais experimentais, a injeção de insulina aumenta em cinco vezes a atividade da G6PD, e esse efeito é amplificado pela coadministração de glicocorticóides [7,60,61]. A indução da atividade enzimática provavelmente depende do aumento da taxa de síntese de proteínas, conforme demonstrado por experimentos de imunotitulação usando anticorpos anti-G6PD [7]. Segue-se que, em alguns casos, a deficiência de insulina pode estar associada à redução da atividade da G6PD. No diabetes induzido por estreptozotocina em camundongos, a atividade da G6PD no fígado foi reduzida em cerca de 50%, especialmente na presença de deficiência concomitante de cobre [38]. Um estudo recente de Parsanathan et al. revelou que o tratamento de células endoteliais aórticas humanas com glicose alta ou palmitato diminui a atividade de G6PD e aumenta citocinas inflamatórias e moléculas de adesão celular, sugerindo outro mecanismo plausível de estado de deficiência de G6PD adquirida [62].
Evidências da literatura, incluindo relatos de casos [33,34,35] e pesquisas clínicas [63], mostraram que a deficiência de G6PD é encontrada com mais frequência em pacientes com diabetes em comparação com a população em geral. No entanto, outros estudos não confirmaram esses achados [64]. Em muitos casos, foi possível demonstrar a coexistência de mutações no gene G6PD [35,65]. No entanto, em casos raros, uma atividade consistentemente menor de G6PD foi descrita em pacientes diabéticos sem uma mutação genética detectável.
Estudos iniciais sugeriram que, em vez do próprio diabetes, é a cetoacidose que desencadeia os episódios hemolíticos, embora apenas na presença de um estado pré-existente de deficiência de G6PD [66,67,68]. Especula-se que a infusão de insulina para corrigir o estado cetoacidótico pode reduzir a disponibilidade de glicose com subsequente depleção de NADPH e capacidade antioxidante prejudicada, afetando especificamente as células β [69]. Mais recentemente, foi proposto que formas adquiridas de deficiência de G6PD no diabetes podem ser precipitadas por descompensação metabólica causada por hiperglicemia grave. Nesses casos, a inibição da enzima não depende da regulação da insulina da síntese de G6PD, mas sim do aumento dos níveis de glicose que promovem a glicação não enzimática da proteína enzimática, fornecendo, portanto, uma explicação mecanicista da perda progressiva da atividade catalítica [70]. Outra contribuição para a inibição da G6PD pode estar relacionada ao aumento do nível de glucagon, que está sempre presente em alguma medida no diabetes, uma vez que se sabe que a injeção de glucagon em animais experimentais reduz a atividade da G6PD [71]. Além disso, as células β pancreáticas são muito vulneráveis ao estresse oxidativo [72], e a depleção da capacidade antioxidante devido a qualquer causa, inata ou adquirida – incluindo deficiência de G6PD – prejudica gravemente a secreção de insulina, criando assim um loop autossustentável.
Uma alta frequência de diminuição da atividade de G6PD, aparentemente não relacionada a mutações genéticas, foi relatada em diabetes com tendência à cetose, uma forma atípica de diabetes comum em descendentes masculinos de africanos subsaarianos, principalmente afro-americanos e afro-caribenhos [36,73]. Um estudo revelou uma alta prevalência (até 42,3%) de deficiência de G6PD sem codificação ou mutações intrônicas de G6PD em pacientes que sofrem dessa forma de diabetes [37], embora o impacto de possíveis problemas técnicos, como armazenamento inadequado ou degradação de amostras, não possa ser totalmente descartado. Curiosamente, a gravidade da deficiência de G6PD foi positivamente correlacionada com a magnitude da deficiência de insulina, confirmando ainda mais que a atividade normal de G6PD é necessária para preservar a função das células β [37]. No entanto, nesta forma de diabetes, é improvável que a deficiência de G6PD seja o único resultado da descompensação metabólica, uma vez que a hiperglicemia induzida experimentalmente por infusão em indivíduos com esta forma de diabetes não inibe a atividade da G6PD nos eritrócitos [36].
2.3.4. Obesity
Adipose tissue displays the highest expression of G6PD after blood [74]. According to some studies, the upregulation of this enzyme in adipocytes is involved in the pathogenesis of metabolic syndrome [74,75]. Increased enzyme activity in fat tissue is usually interpreted as a response to inflammation and oxidative damage that characterizes this condition. However, the enzyme is not increased in the plasma of obese subjects, indicating that its dysregulation is tissue-restricted. Acquired G6PD deficiency has been observed in inflammatory disorders associated with metabolic syndrome. The study by Gheita et al., which recruited 40 cases of rheumatoid arthritis and 30 cases of Sjögren syndrome, reported a frequency of G6PD deficiency of 87.5% and 30%, respectively. In the former case, the level of catalytic activity was significantly lower in patients with concomitant metabolic syndrome [19]. Clearly, these high frequencies cannot be explained by the presence of structural defects of the coding gene and are quite indicative of an acquired form.
2.4. Pré-eclâmpsia
A pré-eclâmpsia indica o aparecimento ou agravamento da hipertensão arterial acompanhada de proteinúria que se desenvolve em mulheres após a 20ª semana de gestação e se apresenta com convulsões não relacionadas a outras causas [76]. O estudo de Afzal-Ahmed et al. forneceu evidências de que as gestações pré-eclâmpticas estão associadas à atividade reduzida de G6PD, depleção de NADPH e comprometimento do equilíbrio redox em eritrócitos e células endoteliais fetais [39]. Nesse distúrbio, a proporção de glutationa reduzida/oxidada é a metade em comparação com mulheres grávidas normotensas [40]. Embora alguns casos de pré-eclâmpsia tenham sido associados a uma mutação no gene G6PD [77], ela deve ser considerada uma forma adquirida de deficiência de G6PD na maioria dos casos.
2.5. Deficiência de micronutrientes
Desde a década de 1950, sabe-se que a atividade da G6PD no fígado diminui em animais submetidos à restrição calórica e aumenta na fase de realimentação, principalmente se a dieta for rica em carboidratos [78,79,80]. Indivíduos humanos com desnutrição protéico-calórica, ou deficiências de micronutrientes específicos, apresentam menor expressão de G6PD, que pode potencialmente ser restaurada por nutrição adequada. Além disso, um estudo realizado em crianças filipinas investigou os níveis de zinco em bebês deficientes e não deficientes em G6PD, encontrando valores significativamente mais baixos (60% vs. 82%) nos primeiros [43]. Esta observação destaca que a deficiência de certos micronutrientes pode ser um fator predisponente para formas adquiridas de deficiência de G6PD que ocorrem em indivíduos desnutridos, embora não se possa excluir que a deficiência de oligoelementos, necessários para responder adequadamente ao desafio oxidativo, possa desmascarar um problema previamente desconhecido deficiência congênita de G6PD.
A deficiência de vitamina D também pode alterar a atividade da G6PD. A forma biologicamente ativa da vitamina D, ou 1,25-hidroxicolecalciferol, é capaz de aumentar os níveis de G6PD interagindo com o receptor da vitamina D (VDR), que é um fator de transcrição que liga um elemento de resposta à vitamina D (VDRE) no primeiro íntron do o gene codificador [81,82]. Em ratos alimentados com uma dieta pobre em vitamina D, a atividade da G6PD parece estar reduzida no fígado e nos rins [42]. No duodeno de ratos com deficiência de vitamina D, a G6PD foi 60% menor do que nos controles [41]. In vitro, a vitamina D é capaz de regular positivamente a G6PD de maneira dependente da dose [81]; in vivo, a deficiência vitamínica pode estar associada à redução da defesa antioxidante devido à depleção da concentração intracelular de glutationa reduzida [31]. Em caso de hemólise inexplicada, o clínico deve suspeitar de deficiência de vitamina D que, se comprovada, deve ser tratada com terapia suplementar.
3. Discussão
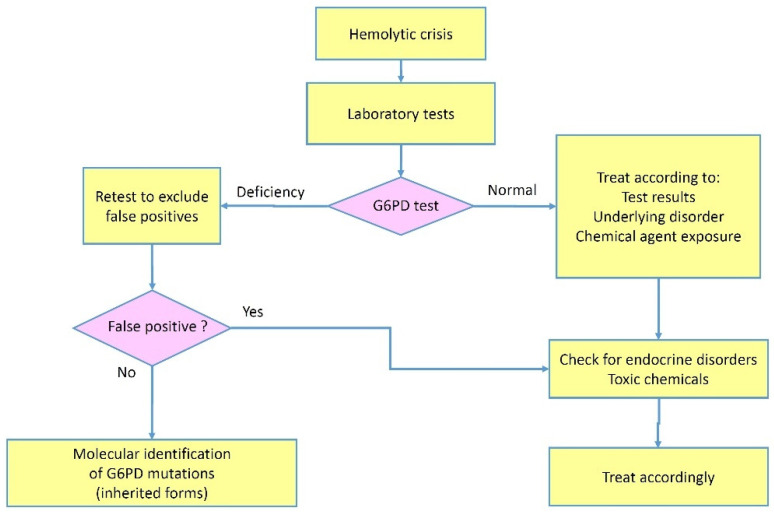
A deficiência de G6PD, uma das enzimopatias mais comuns relatadas em todo o mundo, é geralmente considerada geneticamente determinada por defeitos hereditários no gene G6PD. Embora existam relatos iniciais de formas “adquiridas” na literatura [16,44,48,49], nos quais as mutações genéticas foram totalmente descartadas, essas condições receberam pouca atenção devido à sua raridade e à dificuldade em realizar um diagnóstico preciso. pesquisa de um possível defeito genético. No entanto, a curta duração de suas manifestações clínicas e a falta de evidências de predisposição familiar tornam essas formas nosologicamente diferentes das condições hereditárias usuais. Nos casos mais graves, as formas adquiridas são reveladas por uma crise hemolítica que surge em indivíduos com uma doença subjacente, muitas vezes um desequilíbrio endócrino (diabetes, hiperaldosteronismo primário) ou inflamatório (artrite reumatóide). Em outros casos, a condição é mais branda e não se manifesta com crises hemolíticas, mas pode ser descoberta acidentalmente durante exames laboratoriais de rotina ou para fins de pesquisa. Um cenário típico pode envolver um paciente que desenvolve episódios hemolíticos, nos quais os achados laboratoriais mostram uma redução temporária da atividade catalítica da G6PD que não pode ser confirmada posteriormente. Nesses casos, o distúrbio exibe todas as características de uma condição transitória independente de causas genéticas e razoavelmente atribuível à doença subjacente (Figura 2).
Figura 2
Fluxograma do processo diagnóstico em caso de suspeita de deficiência adquirida de G6PD.
Com base em dados experimentais, os mecanismos envolvidos na deficiência adquirida de G6PD podem ser resumidos da seguinte forma:
A G6PD é fisiologicamente regulada por vários hormônios cuja liberação pode mudar no curso de certas doenças, como hiperaldosteronismo primário, diabetes e hiperglucagonemia, responsáveis pela maioria das formas adquiridas, produzindo uma regulação temporária pós-transcricional da G6PD;
As substâncias químicas podem atuar diretamente como inibidores da G6PD: a ingestão acidental ou voluntária dessas substâncias pode resultar em uma crise hemolítica indistinguível daquela que ocorre nas formas hereditárias;
Em várias doenças hematológicas, postulou-se a presença de inibidores circulantes desconhecidos de G6PD (hormônios? microRNAs?) [46]. Experimentos em que o plasma desses pacientes foi incubado com hemácias de doadores saudáveis mostraram uma diminuição significativa na atividade de muitas enzimas, confirmando assim a presença de um agente inibitório [48]. No entanto, esse mecanismo de inativação foi demonstrável apenas em uma porcentagem limitada de casos.
Uma vez estabelecido o diagnóstico de anemia hemolítica, é importante determinar a causa usando informações da história, exame físico e testes laboratoriais direcionados. De acordo com as causas relatadas de deficiência adquirida de G6PD na literatura, um teste que indique uma atividade de G6PD abaixo da faixa normal requer a exclusão cuidadosa de um teste falso positivo. Em pacientes do sexo feminino, um teste positivo poderia teoricamente resultar da supressão excessiva da inativação do cromossomo X na linhagem sanguínea, especialmente em mulheres idosas. Em geral, um teste qualitativo, por exemplo, um teste de mancha fluorescente sob luz ultravioleta, em nossa opinião, não é recomendável em favor de um teste bioquímico quantitativo. A taxa catalítica deve ser ajustada para os níveis de hemoglobina e contagem de reticulócitos. Quando uma transfusão é planejada, por motivos de urgência, um teste de triagem pode ser usado, mas deve ser seguido por um teste quantitativo confirmatório o mais rápido possível. Se a suspeita de uma forma adquirida de deficiência de G6PD permanecer apesar do teste confirmatório, o teste deve ser repetido pelo menos 120 dias após a resolução do episódio hemolítico (tempo médio necessário para a reposição de hemácias circulantes) para verificar se o estado de deficiência é permanente ou transitório. Testes falso-negativos podem ocorrer devido à co-presença de β-talassemia heterozigótica (frequente em populações mediterrâneas ou em outras populações caracterizadas no passado por malária endêmica). O diagnóstico diferencial deve ser estendido às chamadas anemias hemolíticas adquiridas por diversas causas imunes e não imunes. Essas condições podem exibir todas as marcas de hemólise, mas a G6PD geralmente é normal. No caso de uma doença endócrina subjacente, na qual se sabe que o hormônio envolvido afeta potencialmente a expressão de G6PD, o paciente deve ser tratado, quando possível, corrigindo a endocrinopatia subjacente. Finalmente, na presença de uma deficiência adquirida de G6PD, os médicos devem evitar a administração de medicamentos inseguros que atuam como estressores oxidativos que podem precipitar a hemólise.
Esta revisão pode ter algumas ressalvas. A principal limitação é que até o momento a literatura relatou poucos casos de formas adquiridas de G6PD e, em alguns estudos, a etiologia precisa da condição adquirida não foi identificada. Além disso, é possível que alguns casos tenham sido perdidos pelos autores, tornando a lista incompleta. No entanto, esta revisão narrativa coletou os estudos mais significativos na área, fornecendo pela primeira vez uma visão abrangente da deficiência adquirida de G6PD, que ainda é um problema subestimado, especialmente na prática clínica.
Author Contributions
Conceptualization, G.M.P. and M.P.D.; methodology, G.M.P.; software, G.M.P.; validation, G.M.P. and M.P.D.; formal analysis, G.M.P.; investigation, G.M.P. and M.P.D.; resources, M.P.D.; data curation, M.P.D.; writing—original draft preparation, G.M.P. and M.P.D.; writing—review and editing, G.M.P. and M.P.D.; visualization, G.M.P.; supervision, M.P.D.; project administration, M.P.D.; funding acquisition, M.P.D. All authors have read and agreed to the published version of the manuscript.
References
Leia o artigo original AQUI
Se você chegou até aqui significa que leu o artigo todo na integra, eu não costumo por os artigos completo aqui no blog, geralmente coloco a parte resumida e o link de acesso ao artigo completo, mas esse artigo me chamou tanta a atenção que achei necessário traduzir ele todo e por aqui para que todos tenham o acesso mais rapido.
Obrigado pela leitura e até o próximo artigo.