

Insuficiência hepática em recém-nascidos com deficiência de G6PD
1University of South Carolina School of Medicine, Columbia, SC
2Department of Pediatric Gastroenterology and Hepatology, Levine’s Children Hospital, Charlotte, NC
Correspondence: Milaan Shah, MD (milaan8697@gmail.com).
This is an open access article distributed under the terms of the Creative Commons Attribution-Non Commercial-No Derivatives License 4.0 (CCBY-NC-ND), where it is permissible to download and share the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Abstrato
A deficiência de glicose-6-fosfato desidrogenase (G6PD) é um defeito enzimático comumente herdado que pode se manifestar com hemólise, hiperbilirrubinemia e icterícia e pode causar disfunção renal e hepática. A deficiência de G6PD pode servir como um cofator para doença hepática crônica; no entanto, uma associação com insuficiência hepática não está bem descrita. Apresentamos os casos de 2 recém-nascidos com deficiência de G6PD e insuficiência hepática progressiva resistente ao tratamento com ursodiol que eventualmente necessitaram de transplante de fígado. Nossos casos ressaltam a importância do monitoramento da função hepática em recém-nascidos ictéricos com deficiência subjacente de G6PD e demonstram a potencial precipitação de doença hepática por deficiência de G6PD.
INTRODUÇÃO
A glicose-6-fosfato desidrogenase (G6PD) é uma deficiência genética ligada ao cromossomo X que é o defeito enzimático mais comum no mundo, afetando cerca de 400 milhões de pessoas de etnias principalmente africanas, asiáticas e do Oriente Médio.1 A deficiência de G6PD pode causar o início de hemólise, hiperbilirrubinemia e/ou icterícia em recém-nascidos, mas esses sintomas geralmente podem ser superados com intervenção médica.
A hemólise persistente da deficiência de G6PD pode causar hiperbilirrubinemia grave, que pode precipitar kernicterus, perda auditiva e retardo mental em casos graves.2 No entanto, a hiperbilirrubinemia em neonatos com deficiência de G6PD se deve principalmente à redução da conjugação e excreção hepática, além do aumento da produção de bilirrubina da hemólise.3 A presença de deficiência de G6PD e doença hepática em recém-nascidos é bem descrita, com o mau funcionamento do fígado atribuído à deficiência de G6PD.4 Uma associação entre deficiência de G6PD e insuficiência hepática não está estabelecida, mas a sobreposição de sintomas e etiologia da 2 doenças justifica uma investigação mais aprofundada. Assim, apresentamos 2 casos de neonatos com deficiência de G6PD e insuficiência hepática concomitante.
Caso 1
Um menino afro-americano de 6 semanas de idade, nascido com 36 semanas e 5 dias de gestação, foi observado inicialmente por fezes pigmentadas persistentes e icterícia colestática. Seu trabalho laboratorial inicial revelou níveis elevados de bilirrubina total—6,6 mg/dL, bilirrubina direta—4,1 mg/dL, aspartato aminotransferase (AST)—532 IU/L, alanina aminotransferase (ALT)—244 IU/L e fosfatase alcalina— 593 UI/L e uma gama glutamil transpeptidase normal (GGTP) —25 UI/L. Seu índice normalizado internacional (INR) estava ligeiramente elevado em 1,7, mas normalizou com a suplementação de vitamina K. O hemograma completo estava dentro dos limites normais. Uma investigação extensa para qualquer etiologia hepática primária contribuindo para a icterícia colestática foi negativa.
A avaliação incluiu uma cintilografia hepatobiliar com ácido iminodiacético, que observou excreção biliar e atividade intestinal em 10 minutos, e reação em cadeia da polimerase por citomegalovírus (CMV), que foi observada como levemente positiva com sorologia sugestiva de infecção passada. Um painel de genes de colestase EGL identificou uma variável de significado indeterminado (VUS) no gene CFTR e um VUS nos genes PEX12. No entanto, nenhuma segunda variante foi identificada para nenhum desses genes recessivos. Múltiplas regiões de perda de heterozigosidade foram observadas em seu microarray de polimorfismo de nucleotídeo único. As enzimas lisossômicas e os distúrbios congênitos da glicosilação dos testes de EBV foram normais. Uma biópsia hepática subsequente foi compatível com hepatite neonatal idiopática de células gigantes com colestase e sem fibrose significativa (Figura 1). Uma coloração de CMV revelou um único corpo de inclusão e o vírus Epstein-Barr foi negativo.
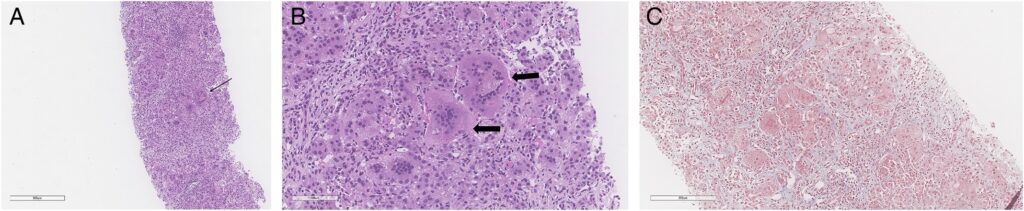
Figura 1.: (A) Visão de baixa ampliação da biópsia hepática mostrando a presença de múltiplas células gigantes. (B) Visão de grande aumento da biópsia hepática mostrando múltiplas células gigantes com colestase, apoptose e um fundo de inflamação mista com linfócitos e eosinófilos. (C) Coloração tricrômica do tecido demonstrando atenuação da fibrose sinusoidal.
Os ácidos biliares séricos e urinários estavam elevados, o que era consistente com doença hepática colestática e disfunção hepática grave, excluindo assim defeitos na síntese de ácidos biliares. Os ácidos orgânicos urinários e os perfis de aminoácidos plasmáticos também eram normais. Posteriormente, todo o sequenciamento do exoma do probando, que foi enviado para genética de prevenção, testou positivo para uma deficiência de G6PD ligada ao X de estado homozigótico/mutação patológica. Digno de nota, a mãe do paciente tinha deficiência de G6PD.
Ao nascer, foi submetido a fototerapia tripla, porém não foi realizada exsanguineotransfusão. Foi iniciado ursodiol 20 mg/kg/dia em doses fracionadas e suplementação vitamínica com ADEKs. Com aproximadamente 6 meses de idade, sua bilirrubina total atingiu um pico de 28 mg/dL, com bilirrubina direta—18,6 mg/dL, AST—1.016 UI/L e ALT—230 UI/L. Seu INR atingiu um pico de 2,8 e não respondeu à vitamina K IV. Sua contagem absoluta de reticulócitos permaneceu elevada em 120 e a hemoglobina caiu para 8,0 mg/dL, exigindo transfusão de concentrado de hemácias. Ele permaneceu hiperamonêmico com um nível de amônia total persistentemente elevado em 120 μmol/L. Ele acabou sendo diagnosticado com insuficiência hepática subaguda e recebeu transplante de fígado nas próximas 2 semanas. No seguimento de 6 meses após o transplante, o paciente estava bem, sem recorrência dos sintomas.
Caso 2
Uma criança recém-nascida afro-americana, nascida com 36 semanas de gestação, apresentou icterícia ao nascer com bilirrubina total—14,1 mg/dL e bilirrubina direta—8,5 mg/dL com 9 horas de vida. Seus testes da função hepática estavam inicialmente elevados com ALT—108 UI/L e AST—262 UI/L, mas sua gama-glutamiltransferase (GGT) estava dentro dos limites normais. Sua hemoglobina era normal em um hemograma completo inicial de 13,5 mg/dL, e sua contagem de reticulócitos estava elevada em 19%. Um teste direto de Coombs foi negativo.
Foi iniciado tratamento com ursodiol, mas as fezes sempre permaneceram amarelo-ouro. Exames adicionais, incluindo ultrassonografia abdominal, ácidos orgânicos na urina, teste de vírus herpes simplex, teste de CMV na urina, receptores solúveis de IL-2 e biópsia hepática, todos retornaram normais. Um painel de colestase genética Emory revelou uma mutação heterozigótica para NOTCH-2 e uma mutação inespecífica em CFTR-5T. Um outro painel de anemia hemolítica para ARUP foi positivo para deficiência de G6PD.
Ela continuou a desenvolver piora da icterícia na idade de 6 semanas com bilirrubina total—37 mg/dL e bilirrubina direta—15,5 mg/dL. Seu INR era 1,9, hemoglobina 9,9 mg/dL, plaquetas 99 e albumina 2,9 g/dL. Ela desenvolveu ascite refratária e necessitou e recebeu um transplante de fígado nas 4 semanas seguintes. Até o momento, ela tem se saído bem.
DISCUSSÃO
A deficiência de G6PD é geralmente uma doença controlável porque a maioria dos indivíduos afetados não apresenta nenhum sintoma negativo, exceto quando são “desencadeados” por infecções virais, certos medicamentos e/ou favas.5 Esses eventos desencadeantes causam dano oxidativo significativo ao glóbulos vermelhos resultando em anemia hemolítica, que é a base para as principais complicações médicas da deficiência de G6PD. Casos graves de deficiência de G6PD podem causar danos aos rins e fígado, mas o mecanismo subjacente ao dano hepático é pouco compreendido.4,6
Nos 2 casos discutidos acima, 2 neonatos apresentaram icterícia colestática crônica que não regrediu apesar do tratamento com ursodiol. Descobriu-se que ambos os bebês tinham hemólise crônica de baixo grau e função hepática comprometida devido à deficiência de G6PD. Digno de nota, em ambos os casos, os recém-nascidos tinham múltiplas mutações únicas em vários genes. O VUS nos genes CFTR e PEX12 e múltiplas perdas de heterozigosidade no polimorfismo de nucleotídeo único no caso #1 e as mutações em NOTCH-2 e CFTR-5T no caso #2 não têm significado clínico conhecido.
Neonatos com deficiência de G6PD podem ter níveis elevados de AST, ALT e INR dentro de 7 dias após o nascimento sem nenhum evento hemolítico precipitante significativo e/ou trauma.4 Isso pode sugerir que a função hepática comprometida é atribuída à deficiência de G6PD. No entanto, a etiologia e a fisiologia subjacentes ao impacto da deficiência de G6PD no fígado não estão estabelecidas e requerem maior elucidação. Se esse mecanismo for melhor compreendido, a investigação da conexão entre as duas condições pode potencialmente revelar causalidade ou a falta dela.
Em conclusão, a deficiência de G6PD pode estar associada à disfunção hepática, servindo como um possível cofator no cenário de insuficiência hepática progressiva/doença hepática crônica. Embora não se saiba se a deficiência de G6PD contribui para o desenvolvimento de insuficiência hepática, nossos 2 casos documentados sugerem que os médicos devem ficar atentos à possibilidade de comprometimento da função hepática como complicação nessa população. Assim, em recém-nascidos com deficiência de G6PD, pode ser particularmente importante monitorar a função hepática quando a icterícia persistir por um longo período de tempo, apesar da intervenção médica. Mais estudos são necessários para entender a relação entre deficiência de G6PD e disfunção hepática em neonatos.
DIVULGAÇÕES
Contribuições dos autores: M. Shah e V. Gopalareddy escreveram e editaram o manuscrito, revisaram a literatura, forneceram as imagens, revisaram o conteúdo intelectual do manuscrito e aprovaram o manuscrito final. M. Shah é fiador do artigo.
Divulgação financeira: Nenhuma a relatar.
Apresentações anteriores: Este caso foi apresentado na Sessão de pôsteres da Associação Americana de Estudantes de Medicina de 2021, em 6 de março de 2021; Virtual e o Dia de Pesquisa da Universidade da Carolina do Sul em 2021, 23 de abril de 2021; Colômbia, SC.
O consentimento informado foi obtido para este relato de caso.
Leia o Artigo original completo AQUI